
Auditorias Internacionais de Fabricantes de IFAs: Nova RDC 301/2019 e CP 683/2019.
Recentemente, com as novas consultas públicas e consequentemente as novas resoluções que irão entrar em vigência, as auditorias internacionais de fabricantes de insumos farmacêuticos, tem sido o foco de vários debates e necessidades da indústria farmacêutica.
A CP 683/2019 é a Consulta Pública que trata da alteração da Resolução – RDC nº 200/2017 (registro de medicamentos), e a Resolução – RDC nº 73/2016, para dispor sobre a submissão do Dossiê de Insumo Farmacêutico Ativo (DIFA) no registro e no pós-registro de medicamento, respectivamente.
Além da CP 683/2019, a ANVISA também publicou as seguintes consultas públicas:
- CP 682/2019: proposta de instituição do Dossiê de Insumo Farmacêutico Ativo (Difa) e da Carta de Adequação de Dossiê de Insumo Farmacêutico Ativo (Cadifa).
- CP 688/2019: critérios para certificação de Boas Práticas de Fabricação para estabelecimentos internacionais fabricantes de insumos farmacêuticos ativos.
- CP 689/2019: proposta de diretrizes de qualificação de fornecedores relacionados ao Regulamento Técnico de Boas Práticas de Distribuição e Fracionamento de Insumos Farmacêuticos, aprovado pela RDC 204/2006.
O objetivo da Agência, além da elevação do nível da regulação de IFA é:
- Harmonização internacional;
- Eficiência processual:
- Processo único: IFA e fabricante de IFA;
- Ganho administrativo;
- Evita decisões diferentes para uma mesma documentação;
- Maior capacidade de análise;
- Maior fluidez aos peticionamentos.
- Isonomia:
- Revogam-se a RDC 57/2009, a IN 15/2009 e a IN 3/2013 (REGISTRO DE IFA);
- Norma única: para o DIFA de todas as submissões de registro de medicamento ou de inclusão ou substituição de local de fabricação do IFA (exceção da transitoriedade).
E o CTD?
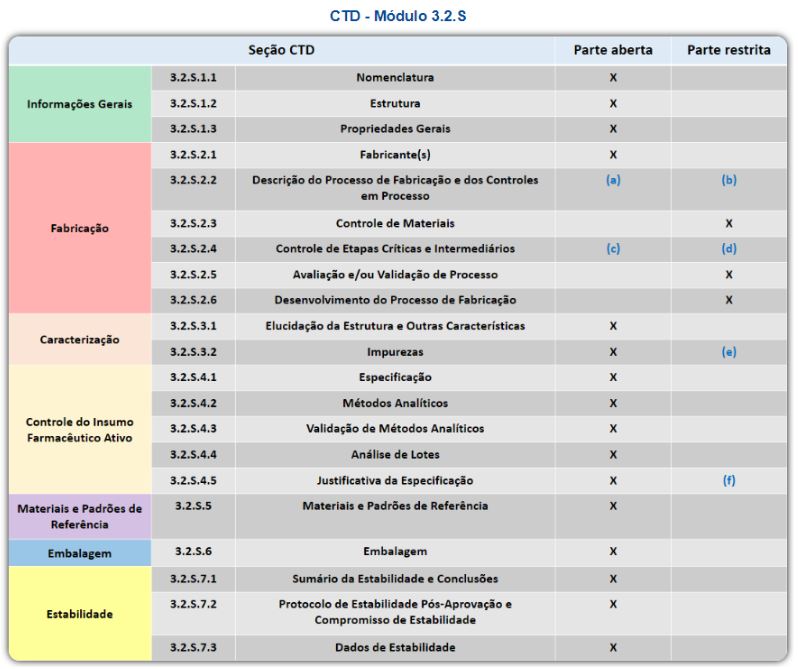
O CTD, ou Documento Técnico Comum, do Inglês Common Technical Document, é um sistema da ANVISA com a finalidade de organização de documentos necessários para o peticionamento do Registro de Medicamentos para uso humano.
A Anvisa ao ser aceita como membro regulador do ICH em novembro de 2016 assumiu o compromisso de implementar os 5 guias nível II, entre eles os Guias ICH M4, que definem a Organização do Documento Técnico Comum para Registro de Medicamentos de Uso Humano. Os Guias ICH M4 definem o formato/estrutura de organização do dossiê de registro e pós-registro de medicamentos dividindo-o em 5 módulos. Esse formato harmonizado revolucionou o processo de análise regulatória e propiciou a implementação de boas práticas de análise dos dossiês.
A organização da informação a ser apresentada em requisições de registro e pós-registro dos seguintes medicamentos:
- Novos
- Inovadores
- Biológicos
- Genéricos
- Similares
- Radiofármacos
- Específicos
- Fitoterápicos.
Novo marco regulatório de IFA

O objetivo desse novo marco regulatório é a harmonização com o PIC/S, adoção dos guias do ICH que tratam da requisitos relacionados à qualidade das IFAs, além da exigência de uma maior nível de qualidade para os fabricantes de IFA, uma vez que o impacto é direto na fabricação de medicamentos em termos de qualidade, risco à saúde do paciente e também regulatório (registro e pós-registro de medicamentos).
Desafios relacionados aos fabricantes de IFAs

Os desafios para adequação desse novo marca regulatório para os fabricantes de IFA, para as indústrias farmacêuticas, até mesmo para a ANVISA é enorme. Dentre eles, a própria Agência destacou os seguintes pontos:
- Sistema: envio de documentos para a Anvisa diretamente do fabricante do IFA (já é um problema e permanecerá caso não se apresente uma solução tecnológica;
- Mudança cultural: comunicação direta com o fabricante do IFA; estabelecimento de responsabilidades de cada ator do processo (fabricante do IFA; detentor do medicamento; Anvisa);
- Vigência da norma: de até 3 anos.
Estes foram os pontos levantados pela ANVISA, mas eu iria além, pois hoje a falta real de conceitos de BPF e de qualidade como um todo de alguns fabricantes já é um fator extremamente preocupante, pois muitas empresas não conseguem compreender a preocupação com o risco à saúde do paciente e fabricam como se fossem indústrias químicas, sem sistema da qualidade, principalmente de higiene, gestão documental, validação e qualificação. Se hoje já é difícil, e muitos fabricantes brasileiros estão sendo interditados, imagino com a publicação da nova RDC?!?
Mas é assim que deve ser. Precisamos de uma regulação mais forte para promover a qualidade das IFAs e consequentemente a qualidade dos medicamentos.
Após o desabafo, retomemos aos fatos…
Registro de IFA pós vigência da norma

Quem não se adequar até o prazo final determinado pela ANVISA será excluído do registro do medicamento.
Por consequência, o registro de medicamento que restar sem fabricante de IFA aprovado terá a fabricação suspensa.
Para aqueles que deixam tudo para a última hora, muito cuidado: o registro de IFA ficará válido até o vencimento não haverá migração automática do registro de IFA para a CADIFA.
Os medicamentos já registrados não há a obrigatoriedade de adoção da CADIFA. Nestes casos, o pós-registro será regido pela CP responsável pela alteração que traz o pós-registro em medicamentos.
Com relação aos novos peticionamentos a partir da vigência e transitoriedade da norma, conforme o Art. 10 da CP 683/2019, não haverá emissão de CADIFA.
Gerenciamento de riscos e as auditorias internacionais em fabricantes de IFAs

Que todos os fabricantes de IFAs devem ser auditados, isso não deve ser questionado, no entanto, é importante avaliar os riscos baseados nos resultados das auditorias internacionais para definir a frequência e o escopo das auditorias. Desta forma o gerenciamento de risco para as auditorias internacionais em fabricantes de IFAs deve:
- Avaliar o compliance da empresa
- Avaliar a disponibilidade do insumo ativo fornecido
- Avaliar o nível da qualidade da empresa
- Avaliar a frequência de compras dos ativos por parte do fabricante do medicamento
Após a mitigação dos riscos por meio da avaliação dos resultados dos possíveis modos de falha, sua consequência (efeito de falha) e as prováveis causas raízes, além da avaliação da severidade x ocorrência x detecção, deve ser avaliado o grau de risco para posterior definição, por parte da equipe do Sistema de Qualidade Farmacêutica, da frequência da realização das auditorias internacionais
Adequação do mercado Farmacêutico

E o mercado farmacêutico está preparado para esta nova demanda?
Pensando tanto em produtos legados quanto em novos projetos há real possibilidade de assumirmos mais esta demanda de trabalho?
Assessoria para execução de auditorias internacionais

Atualmente no Brasil, são poucas as empresas que trabalham com consultorias internacionais, devido à dificuldade em encontrar mão de obra qualificada e devidamente comprovada.
A Consultoria Farmacêuticas possui consultores experts em auditorias de
insumos internacionais, medicamentos e produtos para saúde seguindo os
padrões preconizados pela ANVISA, FDA, ICH e recentemente o PICs.
Nossos consultores possuem experiência comprovada oriunda da indústria
farmacêutica em qualificação de fornecedores internacionais, portanto são
profissionais altamente treinados e acostumados na verificação de compliance
de boas práticas de fabricação (BPF) e também com atuação comprovada em
assuntos regulatórios o que proporciona um diferencial no serviço contratado.
Os profissionais já atuaram em diversos continentes, sendo a China, Índia,
Europa e Estados Unidos.
Diante desta demanda cada vez mais eminente, a Consultoria Farmacêuticas
disponibiliza os seguintes serviços de auditoria internacionais:
Marcação de auditoria – consultoria visando o máximo de aproveitamento em
uma única viagem, diminuindo o custo para a sua empresa.
-Elaboração de agenda robusta de auditoria.
-Avaliação de documentos pré-auditoria como: RPP, DMF, validação de
processo, estabilidade (zona 4b) entre vários outros.
Auditoria in loco, 1 a 2 dias a depender da disponibilidade do fabricante.
-Elaboração de relatório de auditoria
-Elaboração de análise de risco de auditoria
-Acompanhamento e conclusão de CAPA
Os nossos profissionais possuem amplo conhecimento de auditorias
internacionais.
Se a sua empresa tem interesse em auditorias internacionais entre em contato
conosco, pois estamos preparados para oferecer um serviço diferenciado. As auditorias são feitas medicante acordos de confidencialidade, proporcionando a
empresa uma segurança na contratação dos nossos serviços.
Referências
- CP 682/2019: proposta de instituição do Dossiê de Insumo Farmacêutico Ativo (Difa) e da Carta de Adequação de Dossiê de Insumo Farmacêutico Ativo (Cadifa).
- CP 683/2019: proposta de alteração da Resolução da Diretoria Colegiada (RDC) 200/2017 e da RDC 73/2016, para dispor sobre a submissão do Dossiê de Insumo Farmacêutico Ativo (Difa) no registro e no pós-registro de medicamentos, respectivamente.
- CP 688/2019: critérios para certificação de Boas Práticas de Fabricação para estabelecimentos internacionais fabricantes de insumos farmacêuticos ativos.
- CP 689/2019: proposta de diretrizes de qualificação de fornecedores relacionados ao Regulamento Técnico de Boas Práticas de Distribuição e Fracionamento de Insumos Farmacêuticos, aprovado pela RDC 204/2006.
- RDC nº 73/2016: Pós-registro de medicamentos
- CTD ANVISA – GUIA PARA ORGANIZAÇÃO DO DOCUMENTO TÉCNICO COMUM (CTD) PARA O REGISTRO E PÓS-REGISTRO DE MEDICAMENTOS VIGENTE A PARTIR DE 14/08/2019

 na Indústria Farmacêutica: Desafios Críticos e Erros Frequentes no Desenho de Estudos")


