
A elaboração do Manual é resultado do trabalho do Grupo de Trabalho (GT) instituído com a finalidade de contribuir para a adequada implementação da norma. Além do Manual do PATE, foram elaborados PATEs específicos por tipo de mudança para as petições que se enquadram na regra de transição, ou seja, as petições que estão na fila de análise da Anvisa e que podem ser de implementação imediata nos termos da RDC n° 73/2016.
Os modelos disponibilizados são referentes às petições passíveis de mudança, e foram organizados da seguinte forma:
- Anexo I – Item 1: Mudanças relacionadas ao insumo farmacêutico ativo
- Anexo II – Item 4: Mudanças de descrição e composição do medicamento
- Anexo III – Item 5: Mudanças relacionadas ao local de uma ou mais etapas do processo produtivo do medicamento
Grupo de Trabalho PATE
O GT para Proposição de Estratégias e Diretrizes Relativas à Elaboração do PATE teve a participação do gabinete do diretor-presidente, representantes das cinco diretorias da Agência e da área técnica da Gerência de Avaliação de Tecnologia de Pós-Registro de Medicamentos Sintéticos (GEPRE/GGMED). A Diretoria de Autorização e Registros Sanitários (DIARE) coordenou os trabalhos.
Participaram também as associações do setor produtivo que foram convidadas para expor suas dúvidas, dificuldades e sugestões com relação à elaboração do PATE.
Coletadas as informações, o GT realizou análise cuidadosa e profunda das contribuições, que resultaram na atualização do parecer.
Sobre a RDC 73/2016

Qual a finalidade?
Procedimento ordinário: é o procedimento de peticionamento que requer protocolo e que deve aguardar manifestação favorável da Anvisa para a implementação;
Procedimento simplificado: é a simplificação do procedimento ordinário de peticionamento, exclusivamente para as petições que são classificadas como de implementação imediata;
Parecer de Análise Técnica da Empresa (PATE): parecer elaborado pela empresa detentora do registro que aborda no mínimo todos os critérios e documentos previstos na resolução, incluindo uma avaliação crítica de todos os aspectos relevantes para a avaliação da Anvisa. O mesmo deve assegurar que foram realizados e aprovados os critérios e documentos apresentados para a autoridade sanitária com a finalidade de manutenção dos parâmetros de qualidade, segurança e eficácia do produto;
Classificação dos tipos de mudanças

As mudanças de pós-registro são classificadas de acordo com o seu potencial impacto na qualidade, segurança e eficácia do medicamento, podendo ser de implementação imediata, com ou sem protocolo individual, ou depender de aprovação prévia da Anvisa.
- Mudança de implementação imediata
- Mudanças múltiplas concomitantes
- Mudanças múltiplas paralelas
Mudança de implementação imediata
Mudança de pós-registro com potencial impacto significativo na qualidade, segurança e eficácia do medicamento para qual a Anvisa concede autorização prévia para sua imediata implementação pela empresa.
A empresa deve:
- Incluir no HMP ou na petição protocolada individualmente, todas as provas satisfatórias requeridas para a modificação;
- Peticionar segundo o procedimento ordinário, com assunto pertinente, e aguardar manifestação da Anvisa para a sua implementação.
Mudança múltiplas: concomitantes e paralelas
Mudanças múltiplas concomitantes: mudanças decorrentes de uma mudança principal prevista na RDC 73/2016;
Mudanças múltiplas paralelas: duas ou mais mudanças simultâneas e diretamente relacionadas protocoladas conjuntamente;
Principais mudanças a serem protocoladas conforme RDC 73/2016
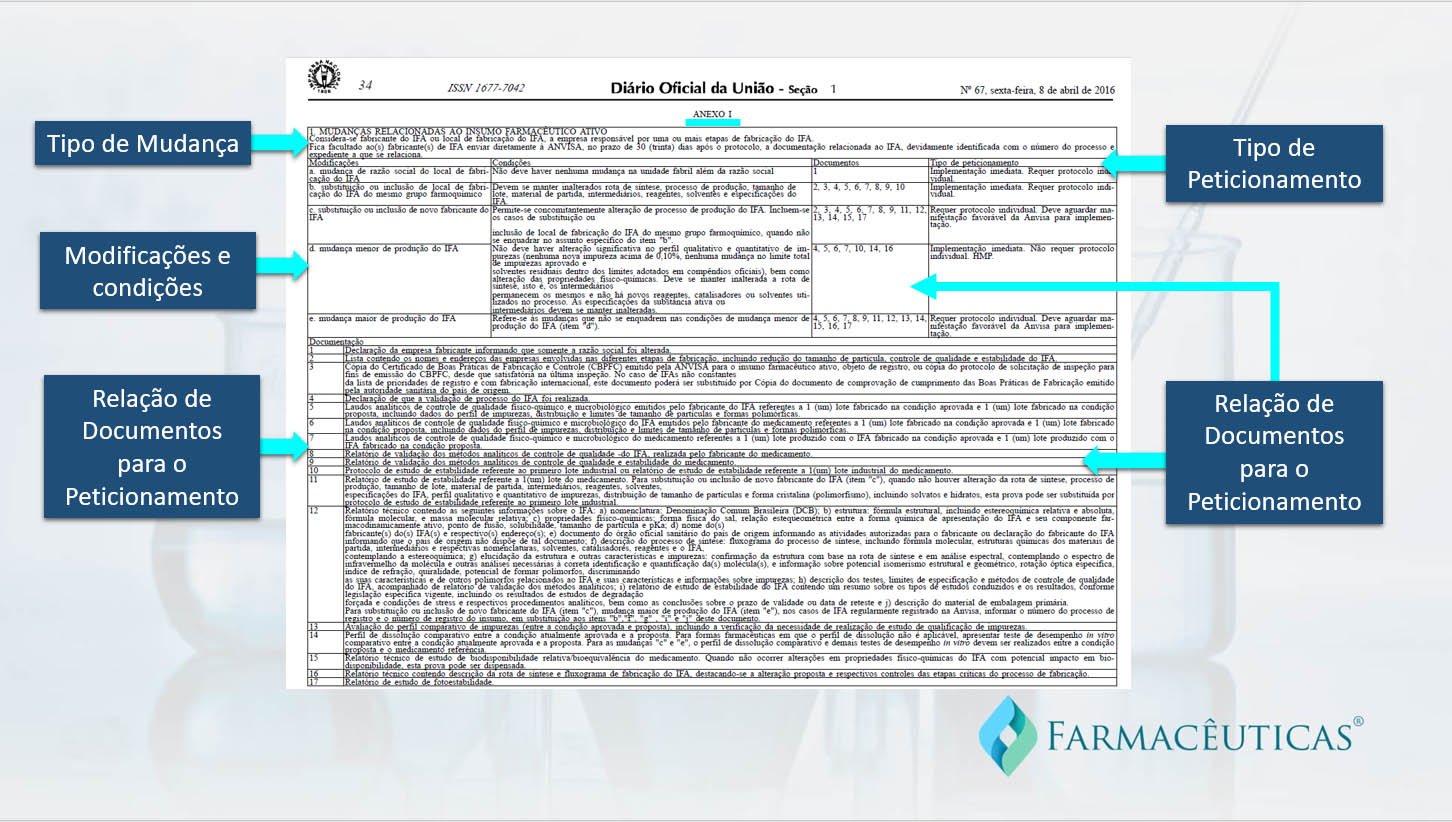
1.MUDANÇAS RELACIONADAS AO INSUMO FARMACÊUTICO ATIVO

a)Mudança de razão social do local de fabricação do IFA
b)Substituição ou inclusão de local de fabricação do IFA do mesmo grupo farmoquímico
c)Substituição ou inclusão de novo fabricante do IFA
d)Mudança menor de produção do IFA
e)Mudança maior de produção do IFA
2. MUDANÇAS RELACIONADAS AOS TESTES, LIMITES DE ESPECIFICAÇÕES E MÉTODOS ANALÍTICOS DO CONTROLE DE QUALIDADE E ESTABILIDADE DO INSUMO FARMACÊUTICO ATIVO E MEDICAMENTO

a)Inclusão de um novo teste
b)Exclusão de um teste ou método obsoleto
c)Estreitamento dos limites de especificação
d)Ampliação dos limites de especificação
e)Mudança menor de método analítico
f)Mudança maior de método analítico
g)Inclusão de método analítico complementar
h)Mudanças realizadas pelo fabricante do IFA
3. MUDANÇAS RELACIONADAS AOS TESTES, LIMITES DE ESPECIFICAÇÕES E MÉTODOS DO CONTROLE DE QUALIDADE DO EXCIPIENTE

a)Mudanças no controle de qualidade do excipiente
4. MUDANÇAS DE DESCRIÇÃO E COMPOSIÇÃO DO MEDICAMENTO

a)Alteração de formato e dimensões de comprimidos, cápsulas, supositórios e óvulos
b)Alteração, exclusão ou inclusão de marcações na forma farmacêutica incluindo marcas em alto e baixo relevo, exceto sulcos e impressões com tinta
c)Alteração ou inclusão de impressão com tinta
d)Mudança menor de sulco
e)Mudança maior de sulco
f)Mudança menor de excipientes para formas farmacêuticas em solução
g)Mudança maior de excipientes para forma farmacêutica em solução
h)Mudança menor de excipientes para formas farmacêuticas semissólidas
i)Mudança maior de excipientes para formas farmacêuticas semissólidas
j)Mudança menor de excipiente para formas farmacêuticas sólidas
k)Mudança maior de excipiente para formas farmacêuticas sólidas
l)Mudança de excipientes para as demais formas farmacêuticas
m)Mudança de excipientes responsáveis pela cor e sabor
n)Inclusão de nova apresentação por alteração de sabor
5. MUDANÇAS RELACIONADAS AO LOCAL DE UMA OU MAIS ETAPAS DO PROCESSO PRODUTIVO DO MEDICAMENTO

a)Alteração de razão social do local de fabricação
b)Inclusão ou substituição de local de embalagem secundária
c)Inclusão ou substituição de local de embalagem primária
d)Inclusão ou substituição de local de fabricação de medicamento de liberação convencional
e)Inclusão ou substituição menor de local de fabricação de medicamento de liberação modificada.
f)Inclusão ou substituição maior de local de fabricação de medicamento de liberação modificada.
g)Inclusão ou substituição de local de fabricação de medicamento estéril
h)Inclusão ou substituição de local de Controle de Qualidade
6. MUDANÇAS RELACIONADAS AO PROCESSO DE PRODUÇÃO DO MEDICAMENTO, EQUIPAMENTO E TAMANHO DE LOTE

a)Mudança menor do processo de produção
b)Mudança maior do processo de produção
c)Inclusão ou substituição de equipamento de embalagem primária
d)Mudança menor de equipamento
e)Mudança maior de equipamento
f)Inclusão menor de tamanho de lote
g)Inclusão maior de tamanho do lote
7. MUDANÇAS RELACIONADAS À EMBALAGEM DO MEDICAMENTO

a)Mudança menor de composição de embalagem primária
b)Mudança maior de composição de embalagem primária
c)Mudança menor da forma e dimensões da embalagem primária
d)Mudança maior da forma e dimensões da embalagem primária
e)Mudança de parte da embalagem primária sem contato com o medicamento
f)Mudança menor de embalagem secundária ou envoltório intermediário
g)Mudança maior de embalagem secundária ou envoltório intermediário
h)Mudança relacionada ao material inerte
i)Mudança relacionada ao diluente
j)Mudança relacionada ao acessório
k)Mudança relacionada ao controle de qualidade da embalagem
l)Inclusão de novo tipo de embalagem primária
8. MUDANÇAS RELACIONADAS À NOVA APRESENTAÇÃO DO MEDICAMENTO
a)Inclusão de nova apresentação
9. MUDANÇAS RELACIONADAS AO PRAZO DE VALIDADE OU AOS CUIDADOS DE CONSERVAÇÃO DO MEDICAMENTO
a)Redução do prazo de validade
b)Ampliação do prazo de validade
c)Mudança dos cuidados de conservação
d)Mudança de condição de armazenamento adicional
10. INCLUSÃO DE NOVA CONCENTRAÇÃO
a)Inclusão de nova concentração para medicamentos novos
b)Inclusão de nova concentração para medicamentos genéricos e similares
11. MUDANÇAS RELACIONADAS À POSOLOGIA, AMPLIAÇÃO DE USO, INCLUSÃO DE NOVA VIA DE ADMINISTRAÇÃO, NOVA INDICAÇÃO TERAPÊUTICA
a)Inclusão de nova posologia para medicamentos novos
b)Ampliação de uso para medicamentos novos
c)Inclusão de nova via de administração para medicamentos novos
d)Inclusão de nova indicação terapêutica para medicamentos novos
12. MUDANÇAS RELACIONADAS AO NOME DO MEDICAMENTO, CANCELAMENTO DO REGISTRO DO MEDICAMENTO e EXCLUSÃO DE LOCAL DE FABRICAÇÃO DO FÁRMACO, LOCAL DE EMBALAGEM PRIMÁRIA, LOCAL DE EMBALAGEM SECUNDÁRIA E/OU LOCAL DE FABRICAÇÃO DO PRODUTO

a)Mudança de nome comercial do medicamento
b)Cancelamento de registro da apresentação
c)Cancelamento de registro (todas as apresentações)
d)Exclusão de local de fabricação do fármaco, local de embalagem primária, local de embalagem secundária e/ou local de fabricação do produto
Relação de documentos em caso de mudança

A relação de documentos é assustadora, mas vou ajudar fazendo uma lista do que seria “o básico”:
- Change request ou Formulário de Controle de Mudanças
- Análise de risco
- Protocolo de validação de processo, e /ou relatório sumário de validação
- Protocolo de estudo de estabilidade
- HMP
- PATE
- Protocolização eletrônica
E o que deve conter no HMP?
- Todas as mudanças pós-registro de implementação imediata, com ou sem protocolo, bem como as que tiveram aprovação prévia da Anvisa;
- Informações complementares, incluindo:
a) a lista de lotes fabricados ou importados no ano, destinados exclusivamente à comercialização no mercado brasileiro, incluindo data de fabricação, número e tamanho do lote (massa/volume e unidades farmacotécnicas);
b) última versão do(s) documento(s) contendo testes, limites de especificação e métodos analíticos de controle de qualidade do medicamento, conforme aprovado;
c) relatórios de estudos de estabilidade de acompanhamento concluídos e de validação de processo.
d) demais informações que não são caracterizadas como mudanças pós-registro, mas que são atualizações de informações apresentadas no registro.
- O HMP deve estar atualizado e facilmente disponível na empresa para apresentação à autoridade sanitária quando requerido.
- Os dados do HMP deverão ser protocolados anualmente, no mês do aniversário do registro do medicamento, mesmo não havendo nenhuma mudança pós-registro, e deverão ser referentes ao período de 12 (doze) meses anteriores ao seu protocolo.
- O protocolo do HMP deve ser realizado através do peticionamento eletrônico e selecionada a modalidade de petição eletrônica, não havendo a necessidade de envio da documentação em papel.
Dependendo da classificação da mudança, a relação de documentos solicitados pela Anvisa pode variar, mas já foram previamente definidas na RDC 73/2016, conforme demonstrado abaixo:
Sobre o PATE
O PATE é um documento a ser elaborado antes da implementação da mudança pós- registro. Deve ser submetido à ANVISA na petição pós-registro de protocolo individual e na mudança reportada via HMP, conforme o caso, e pode ser solicitado no momento de uma inspeção de pós-registro.
- Nos casos em que não ocorrerem mudanças dentro do prazo anual que é contemplado o HMP, não é necessário incluir o PATE a este HMP.
- Deve ser impresso e em mídia eletrônica.
- Gestão documental: Número sequencial para cada processo com versão, páginas numeradas, histórico – deverá ser estabelecido o expediente referente a cada PATE e a qual(is) forma(s) farmacêutica(s), concentração(ões) e apresentação(ões) este expediente/ peticionamento se refere.
- Deve ser utilizado como uma ferramenta da qualidade.
- Deve ser elaborado segundo a mudança – destacada
- Destacar as informações mais relevantes
- Conclusões sobre as provas técnicas e os riscos envolvidos na mudança.
- A avaliação crítica a ser realizada pela empresa deverá abordar:a) Elementos técnicosb) Legislação sanitária e os guias vigentes da ANVISA, e na ausência destes, guias internacionais, que sustentaram a decisão para a realização e a implementação da mudança pós-registro no medicamento.Para os casos de pós-registros em que não haja enquadramento descrito na legislação vigente, após definição por parte da ANVISA, a empresa deverá apresentar no PATE o racional para a mudança proposta e a justificativa para as provas apresentadas.
Mudanças paralelas
A empresa deverá redigir um único PATE contemplando todas as mudanças, incluindo as concomitantes.
Responsáveis pela aprovação dos gestores:
- Responsável técnico
- Garantia da qualidade
- Assuntos regulatórios
- Demais responsáveis pela mudança realizada.
Por onde começar em caso de mudança?

1. Abrir um controle de mudanças
2. Elaborar uma análise de risco para identificar os possíveis riscos e impactos de acordo com a RDC nº 73/2016 e classificação da mudança – Anexar ao PATE
3. Classificar a mudança
4. Elaborar o Histórico de Mudanças do Produto (HMP) anualmente – protocolar no mês do aniversário do registro do medicamento, mesmo não havendo nenhuma mudança pós-registro, devendo ser referente ao período de 12 meses anteriores ao seu protocolo.
5. Incluir as provas necessárias ao HMP
6. Elaborar o PATE e anexar ao HMP
7. Mudança imediata: Peticionadar segundo o procedimento ordinário e assunto
8. Aguardar manifestação da Anvisa para a sua implementação
9. Mudança ratificada ou indefirida
10. Em caso de indeferimento, as condições anteriores à mudança deverão ser restabelecidas imediatamente após a manifestação da Anvisa ou a fabricação do medicamento deverá ser temporariamente descontinuada.
11. Aprovação: Publicação no Diário Oficial da União, ou em outro meio de divulgação institucional – a empresa terá até 180 dias para implementação da modificação, exceto quando houver manifestação contrária da Anvisa.
12. Após a produção do primeiro lote com a mudança aprovada, não será permitida a produção de lotes em condição diferente.
Para mudanças paralelas:
- A empresa deve protocolar cada mudança individual apresentando documentação única que contemple todas as provas relativas a cada um dos assuntos de petição.
- Fazer a descrição das alterações paralelas e sua correlação que devem constar na justificativa a que se refere o artigo 15, inciso III, da Resolução.
- Apresentar a avaliação do efeito aditivo de mudanças individuais paralelas no que se refere ao potencial impacto na qualidade, segurança e eficácia do medicamento e apresentar as provas adicionais, quando necessário.
Para mudanças concomitantes:
- Nos casos de mudanças concomitantes, o peticionamento deve ser referente à mudança principal e a informação sobre a mudança concomitante deve ser descrita na justificativa.
- As únicas mudanças que serão consideradas como concomitantes são aquelas explícitas na norma.
- Devem ser apresentadas as provas relativas a todas as mudanças.
- Quando a documentação solicitada em mudanças concomitantes for divergente, deverá ser apresentada a documentação relativa à mudança principal.
Conclusão
A implantação da RDC 73/2016 não está sendo nada fácil.
Exige uma comunicação e alinhamento de informações grande entre as equipes que atuam na área industrial (garantia, validação, produção, controle, farmacotécnico, etc) e de assuntos regulatórios, algo realmente inédito até a publicação da norma. Vamos ser sinceros!!!
Além disso, o volume de trabalho que gerou, e quantidade de documentos que precisam ser elaborados, é enorme. Isso sem falar o quanto a norma ENGESSOU a produção e até as empresas fabricantes de medicamentos de forma geral.
Até entendo a necessidade de tais controles, mas não foram em excesso?
Veja o exemplo a questão do fornecedor de IFA, a empresa fabricante de medicamentos agora deve ficar limitado a produzir com apenas um único fornecedor de um ativo específico por produto? E se houver problemas na importação/exportação do ativo?
Ou mesmo como você terá tanta certeza de que ele não fará alteração na rota de síntese entre uma auditoria internacional e outra que a empresa fará?
O fornecedor vai comunicar a empresa tais alterações?
Sinceramente, duvido muito.
Enfim, a vigência só está começando, ainda vamos ter muito o que falar a respeito.
Sendo assim, vamos aguardar as cenas dos próximos capítulos…

 na Indústria Farmacêutica: Desafios Críticos e Erros Frequentes no Desenho de Estudos")



[…] 11, 2016 A RDC 73/2016 entrou em vigor ainda esta semana e o resultado da atuação da ANVISA já pode ser notado. […]
[…] para a divulgação deste tema, visto as últimas diretrizes da ANVISA com as publicações das RDC nº 73/2016 e RDC n° 76/2016 que fala sobre o processo de protocolização de mudanças que impactam […]
[…] já é realizado atualmente. Porém, a distribuição das petições protocoladas conforme a RDC nº 73/20016 será iniciada com a vigência da norma, com a finalidade de acompanhamento da implementação […]
Oi Fernanda, boa noite!
Primeiramente parabéns pelos esclarecimentos da RDC 73 e Pate, realmente ficou muito bom.
Estou preparando uma apresentação da RDC 73 para treinar as outras áreas da empresa impactadas com esta nova norma e fiquei com uma dúvida.
Pq vc afirma que as empresas deverão fabricantes de medicamento deverão ter aprnas um fabricante aprovado de IFA por produto. Onde vc viu isto? Como concluiu isto?
Obrigada
Oi, Priscilla!
Na verdade, as empresas não precisam ter um único fornecedor de ativo, mas é algo que se tornou muito complicado e caro.
A questão é que o fornecedor tem que ser qualificado e o registro do produto deve considerar o fabricante do ativo. Caso haja alteração ou inclusão, a mudança deve ser peticionada na ANVISA e aguardar deferimento para a fabricação com novo fornecedor.
Lembrando que o seu processo deve ser validado para cada fornecedor de ativo. Caso mude agora terá que fazer nova validação.
Sendo assim, se tiver 2 fornecedores para a mesma IFA, terá que acompanhar 3 lotes para validação de cada fornecedor (total de 6 lotes). Antes não era necessário.
É por isso, que a tendência no mercado é de ter um ativo mono source.
Espero ter esclarecido.
Abs