
Há muito tempo, diria até mesmo anos, a Anvisa tem cobrado das indústrias farmacêuticas estudos para os produtos/insumos com resultados fora da especificação (FDE), ou como é mais conhecido OOS – Out Of Specification. No entato, a RDC nº 17/2010 não trazia muitos esclarecimentos de como conduzir as investigações e muito menos dava diretrizes sobre a correta elaboração do relatório, apenas exigia que deveria ser feito e verificava os documentos durante a inspeção.
Sem suporte técnico até então, a saída das indústrias era a consulta ao guia do FDA de 2006 Investigating Out-of-Specification (OOS) Test Results for Pharmaceutical Production.
Porém, recentemente a Anvisa lancou o Guia nº 8/2017 – Investigação de Resultados Fora da Especificação (FDE), padronizando a forma de como executar as investigações, em caso de resultados fora da especificação, e dando uma luz a quem estava perdido neste sentido.
E para que entenda um pouco mais sobre o que seria o FDE ou OOS, ou até mesmo para que conheça um pouco mais do guia, trouxemos algumas informações relevantes aqui neste post:
O que são resultados fora da especificação?

Um resultado de análise é considerado fora de especificação caso o valor obtido esteja acima ou abaixo do resultado esperado (especificação), mesmo que o método de análise utilizado para o teste seja padronizado, farmacopeico e/ou validado.
O resultado fora da especificação pode ser obtido durante uma análise executada no Controle de Qualidade (físico-químico e microbiológico), ou durante o controle em processo do produto.
Qual a diferença entre o FDE e OOS?

O guia FDE elaborado pela ANVISA traz mais conteúdo/informações de como proceder com as investigações, além de se apresentar fluxogramas para explicar de maneira mais segura e objetiva as decisões a serem tomadas na investigação dos resultados fora da especificação durante os testes laboratoriais.
O guia OOS foi elaborado pelo FDA, sendo mais obsoleto. Este guia define a investigação em apenas duas fases, a Fase I e II. Já o guia FDE consiste em uma versão mais robusta, possui várias fases para tomada de decisões durante a investigação: Fase I, Fase Ia, Fase Ib, Fase II e Fase III.
Para que serve o Guia FDE?

Este guia fornece às indústrias do ramo farmacêutico orientações gerais sobre o processo de avaliação de resultados de ensaios analíticos físico-químicos e microbiológicos, quando aplicáveis.
Deste modo, o guia pode ser aplicado em testes laboratoriais que são realizados em:
- Materiais de partida
- Matérias-primas
- Produtos intermediários
- Insumos farmacêuticos ativos
- Excipientes
- Materiais de embalagem
- Produtos acabados
- Testes de estabilidade de produtos
Os testes efetuados em laboratório devem ser confiáveis e adequados às condições de uso, além de serem validados e/ou terem a adequabilidade confirmada quando se tratarem de métodos compendiais.
Os resultados FDE/OOS também podem ser detectados durante os testes de controle em processo.
[quote_center]Os produtos fora das especificações não devem ser liberados para a comercialização ou uso.[/quote_center]
[quote_box_center]Nota: não é só a indústria farmacêutica que deve proceder com este estudo investigacional. Todo fabricante de cosméticos e produtos para saúde que exportarem seus produtos para a Europa e Estados Unidos devem possuir, obrigatoriamente, procedimentos aprovados sobre a condução correta das investigações para os casos de OOS/FDE, além dos relatórios devidamente preenchidos.[/quote_box_center]
O que fazer em casos de resultados fora da especificação?

Quado os resultados fora de especificação (FDE) forem detectados estes devem ser devidamente investigados e os registros obtidos devem conter informações organizadas e detalhadas das etapas de investigação.
Neste contexto, a investigação de resultados fora de especificação deve ser aplicável a:
- Testes para liberação de matérias-primas, materiais de embalagem, intermediários, materiais de partida, produtos a granel e terminados de medicamentos, insumos farmacêuticos ativos (IFA), e excipientes farmacêuticos;
- Testes de controle em processo – se utilizados para cálculo de rendimento ou para decisão sobre o lote.
[quote_box_center]Resultados de testes de controle em processo obtidos durante o ajuste de equipamentos e dispositivos e que não atenderam às especificações não são considerados como FDE;[/quote_box_center]
- Resultados de estudos de estabilidade de produtos terminados de medicamentos, insumos farmacêuticos ativos (IFA) e excipientes farmacêuticos;
- Resultados fora de especificação em amostras de retenção de lotes (testados em decorrência de investigação).
É importante ressaltar que mesmo que seja adotada uma política de sempre rejeitar um lote com um resultado FDE, a abertura de uma investigação é necessária para se determinar se o resultado está associado com outros lotes do mesmo produto ou mesmo de produtos diferentes. Assim como, para investigar e determinar as possíveis causas raízes do evento, de forma a assegurar que não haja reincidência do mesmo erro ou deficiência.
A investigação deve ser realizada de forma aprofundada, imparcial, bem documentada, com embasamento técnico e ser iniciada imediatamente após a obtenção de um resultado não esperado e sem explicação aparente. Normalmente a investigação é dividida em fases.
Fases da Investigação dos resultados FDE/OOS

O primeiro fluxograma demonstra como promover a investigação de FDE:
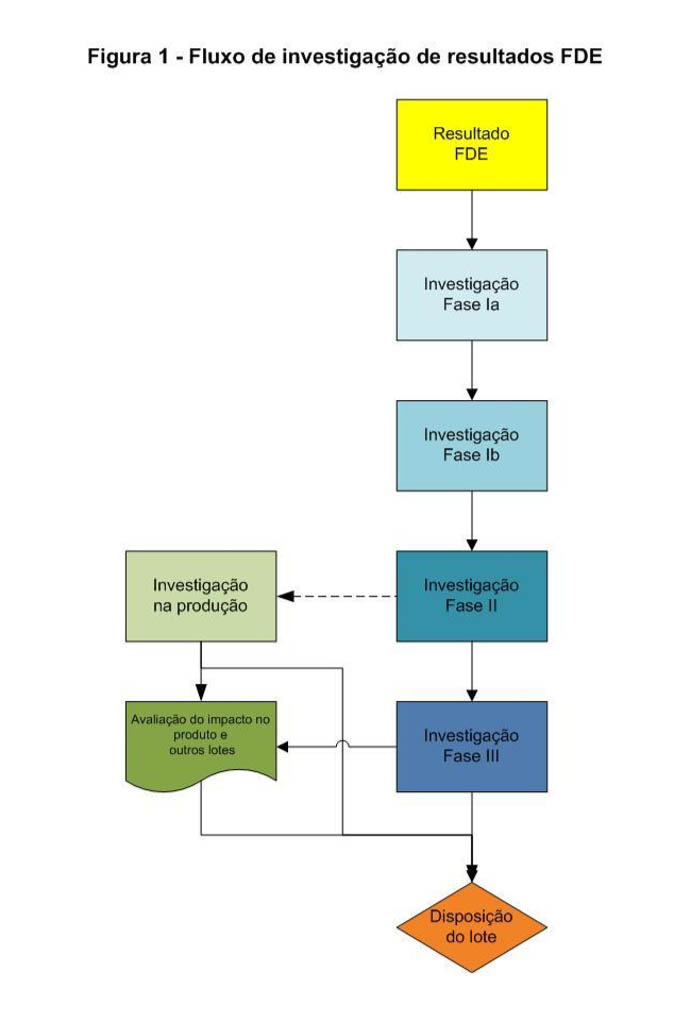
Fase I
Avalia a exatidão dos dados obtidos no laboratório e pode ser dividida em Fase Ia e Fase Ib.
Fase Ia
É a fase de investigação na qual, sob responsabilidade do analista, se verifica a ocorrência de algum erro óbvio no laboratório. Geralmente os erros óbvios são relacionados a circunstâncias externas tais como:
- Queda de energia;
- Falha no equipamento;
- Erros detectados pelo analista antes da conclusão da análise;
- Erros na execução dos testes como o derramamento de amostras;
- Crescimento de microrganismos em placas de Petri armazenadas fora de locais apropriados Falta de controles positivos ou negativos são erros óbvios.
A causa do erro não sendo encontrada, a investigação deve prosseguir para a Fase Ib.
Fluxograma 2 mostra como proceder neste caso:
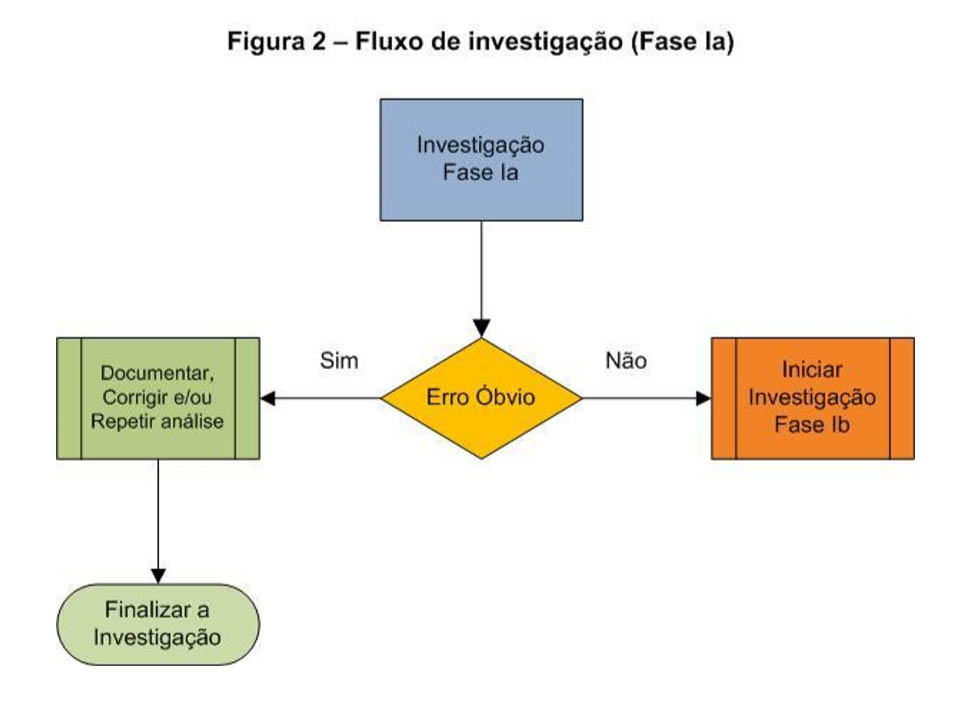
Fase Ib
Deve ser realizada com a participação do analista e seu supervisor. Nesta fase a investigação deve ser focada exclusivamente na revisão dos dados brutos, nas informações sobre os equipamentos, vidrarias, procedimentos e métodos de análises envolvidos. Normalmente utiliza-se uma lista de verificação (check-list) como guia.
Nesta fase, no mínimo, as seguintes informações devem ser revisadas:
- O método analítico executado correspondente à última versão aprovada;
- A validação das metodologias analíticas;
- O procedimento de amostragem e os registros de tomada de amostras. A verificação deve se ater a correta informação sobre as quantidades, a identificação, a guarda e a correta localização de armazenamento e tomada das amostras;
- A integridade das amostras relacionada à estanqueidade do fechamento dos recipientes e ao seu armazenamento temporário;
- Os registros de preparação de meios de cultura e reagentes;
- Os prazos de validade de soluções, meios de cultura e reagentes;
- A quantidade de passagens/gerações e a rastreabilidade das cepas de microrganismos padrões;
- Os dados brutos encontrados em todas as etapas da análise, por exemplo, cromatogramas e espectrogramas. A verificação deve buscar a identificação de dados anormais ou suspeitos;
- Os cálculos derivados dos dados brutos e a integridade dos dados. Deve-se incluir nessa verificação a ausência de alterações indevidas nos dados registrados pelos sistemas automatizados (integridade dos dados);
- As calibrações dos instrumentos, seus registros e se os procedimentos de calibração foram realizados apropriadamente;
- As qualificações e os registros de uso de equipamentos utilizados na preparação, encubação e esterilização;
- Os livros de registros dos equipamentos e confirmação do desempenho e o uso dos instrumentos designados no procedimento de análise;
- A adequabilidade do método (adequabilidade do sistema);
- O treinamento do analista no método;
- O desempenho do método, ou seja, a concordância do resultado encontrado com o resultado esperado, com base nos dados de validação e dados históricos;
- Os dados de identificação e de qualidade dos padrões de referência, padrões de trabalho, soluções reagentes, solventes e outras substâncias utilizadas. A verificação deve buscar dados da validade, especificações, aparência, condições de armazenamento e qualquer outra de suas especificações de controle de qualidade;
- Limpeza e armazenagem correta dos recipientes, vidrarias e utensílios utilizados na amostragem e na análise;
- Indícios de contaminação da amostra. Por exemplo: a amostra permaneceu aberta ou abandonada; o sistema de insuflamento e exaustão são adequados e estavam funcionando corretamente no momento da amostragem e se houve compartilhamento de utensílios;
- Histórico de problemas relacionados ao ensaio em questão;
- Desvios das condições ambientais relacionadas à temperatura, umidade ou incidência luminosa durante o ensaio;
- Verificação dos dados de lotes que estavam sendo analisados em conjunto;
- Outras atividades que ocorreram durante o teste e que poderiam interferir no resultado.
Condições adicionais devem ser consideradas e investigadas caso o teste fora da especificação seja microbiológico.
Assim, deve-se verificar:
- Se a aparência do meio de cultura nas placas amostrais utilizadas está de acordo com o esperado;
- A localização ou disposição das colônias nas placas e nas áreas de contato amostradas;
- Se o meio de cultura está uniforme e íntegro sobre a placa ou há rachaduras ou outros sinais de degradação;
- A ocorrência de contaminação em outras amostras ou em outros testes realizados na sequência do teste que disparou o evento investigado. Deve-se considerar inclusive os resultados de monitoramento ambiental no período considerado;
- Se os controles negativos e positivos estão de acordo com o esperado;
- Se os meios de culturas e/ou reagentes utilizados estavam corretos e a sua armazenagem antes do uso;
- A integridade do recipiente contendo a amostra;
- As condições de armazenamento da amostra desde a amostragem até o uso no teste;
- Se o tempo entre a amostragem e a realização do teste estava dentro do tempo suportado pelos estudos prévios;
- Se as condições de incubação foram satisfatórias; e
- Se o microrganismo isolado e identificado, motivo do resultado fora de especificação, pode auxiliar na investigação da causa raiz.
Nesta fase, é indicado tirar fotos das amostras e dos materiais após a leitura dos resultados dos testes.
A entrevista com o analista também é indicado.
Caso tenha sido evidenciado um erro no laboratório, os resultados FDE devem ser invalidados.
Em caso contrário, ou seja, quando não haja evidência clara de erro laboratorial, deve se iniciar uma investigação ampla incluindo a área de produção tal como descrito na Fase II.
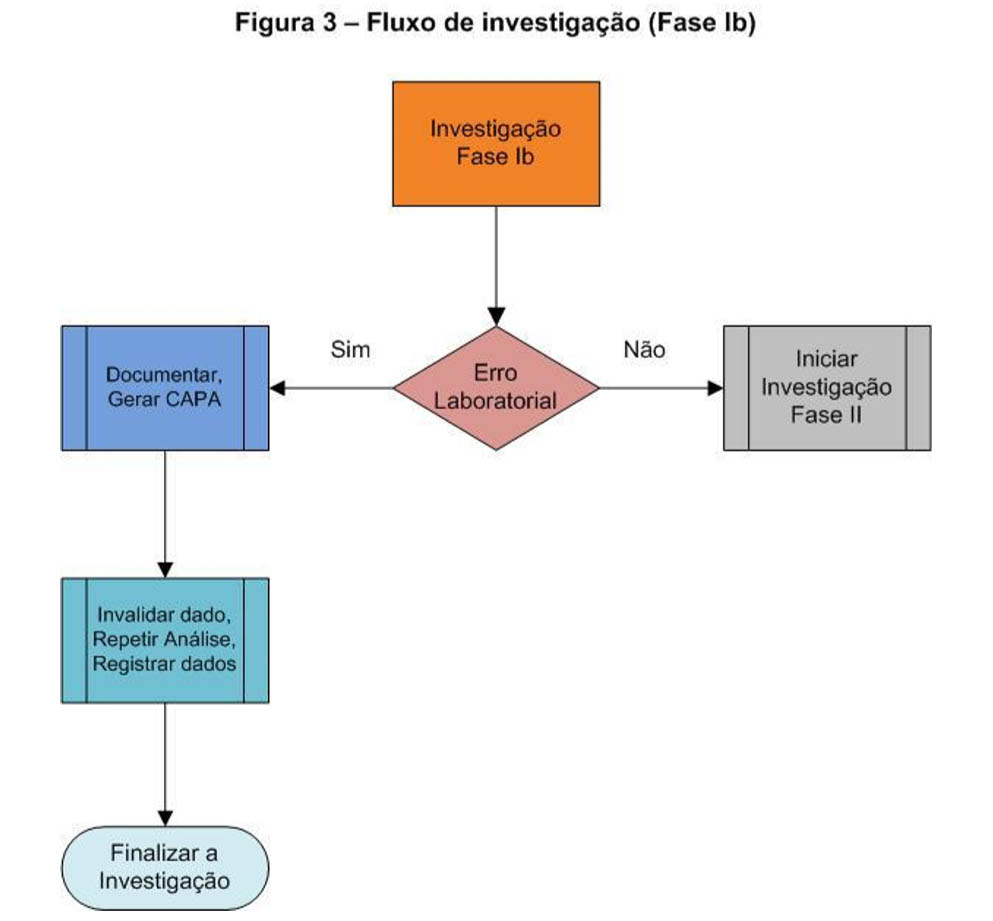
[quote_box_center]Nota: Para quem não sabe, quando o guia fala em CAPA, não é a capa do relatório de investiggação, mas sim o desvio. Isso porque CAPA vem do termo em inglês CORRECTIVE ACTION PREVENTIVE ACTION.[/quote_box_center]
Fica a dica ; )
Fase II
Inicia-se a fase II quando a investigação na fase I efetuada no laboratório, não é capaz de identificar a causa provável do resultado FDE.
Deve investigar se a causa raiz do resultado FDE não está associada à produção, mas pode ser estendida ao almoxarifado, validação ou outra área envolvida. Na investigação relacionada com a área de produção, o enfoque deve se voltar para a revisão dos procedimentos e registros de produção e de amostragem, além de incluir a possibilidade de realizar testes de laboratório adicionais para comprovação de hipóteses.
A avaliação do impacto do resultado FDE em lotes já distribuídos também deve ser iniciada neste momento.
Toda a investigação deve ser totalmente registrada e documentada. Ou seja, toda documentação deve ser revisada, os testes efetuados, os registros avaliados. Tomadas de decisões e análises de risco devem fazer parte da documentação de suporte.
Fluxograma de investigação da fase II, incluindo investigação na produção:
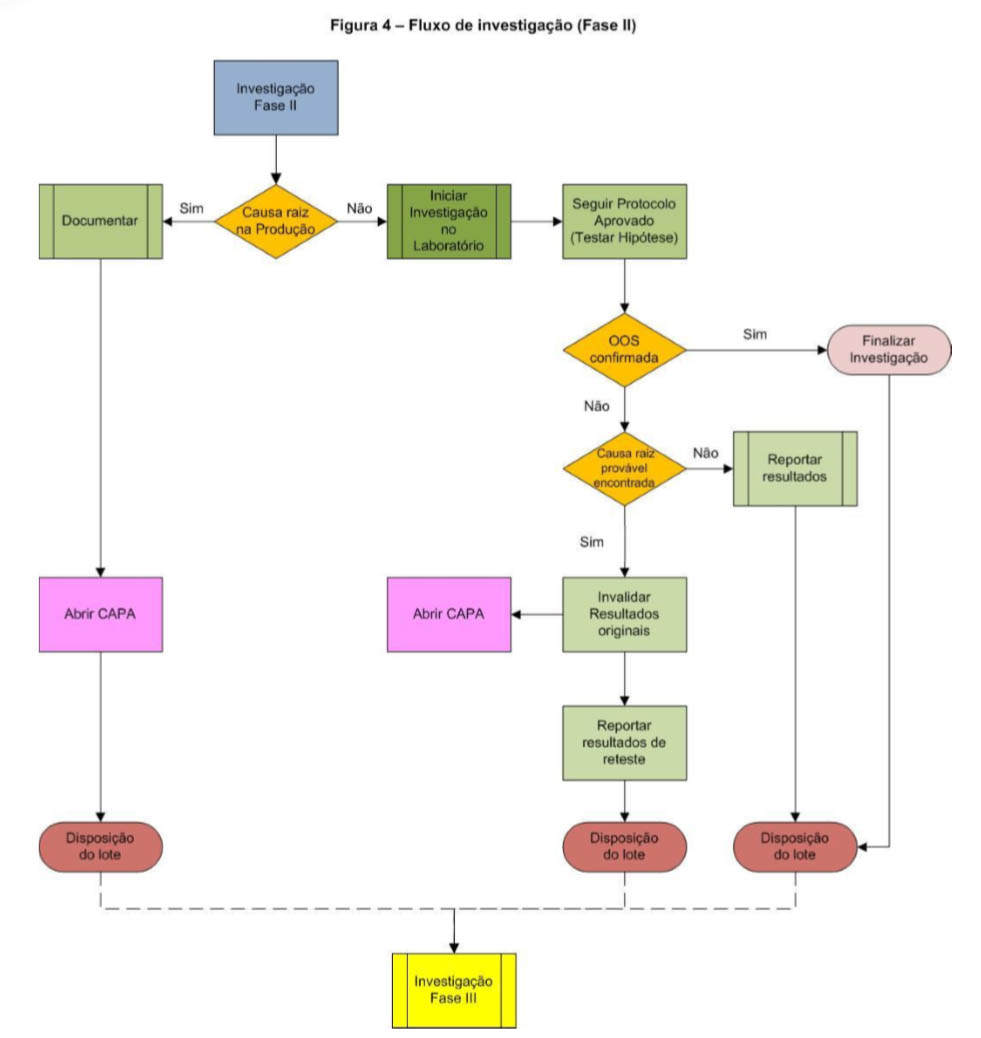
Revisão da Produção

A investigação dessa fase deve ser efetuada pela Garantia da Qualidade e envolver outros departamentos implicados como de produção, desenvolvimento, manutenção e engenharia. Em casos onde diferentes etapas de fabricação são realizadas por terceiros, todas empresas potencialmente envolvidas devem ser incluídas na investigação.
A investigação da Fase II deve ser finalizada em um tempo pré-definido, além de ser aprofundada, detalhada, imparcial; bem documentada e tecnicamente embasada.
O relatório final da investigação deve incluir:
- O motivo da investigação claramente descrito;
- Um sumário dos aspectos do processo de produção que podem ter causado o problema;
- O resultado da revisão da documentação de produção com a atribuição da provável causa (hipótese);
- Os resultados da revisão efetuada para determinar se o problema já ocorreu previamente e a descrição das ações corretivas e preventivas que foram tomadas na ocasião do fato.
Caso a investigação nessa fase confirme o resultado FDE e a causa raiz for determinada, a investigação pode ser finalizada e o produto ter sua disposição final definida.
Entretanto, a investigação a outros lotes que possam estar associados com a causa raiz deve ser finalizada. O reprocessamento de materiais deve obedecer às condições dispostas nas diretrizes de boas práticas de fabricação.
A determinação da causa raiz pode evidenciar inconsistência no processo de fabricação, incluindo problemas com a formulação, validação de processo, controles de qualidade entre outros. Em todos os casos, deve ser avaliado o impacto na consistência do processo produtivo e a avaliação sobre a necessidade de revisar sistemas, estudos de validação e controles necessários.
Ações corretivas e preventivas devem ser propostas, aprovadas, implementadas e monitoradas.
Testes laboratoriais adicionais

Podem ser realizados testes laboratoriais adicionais por meio de reteste da amostra original ou reamostragem concomitantemente ou após a investigação na produção.
Somente em caso de averiguações sobre uma hipótese documentada da causa do resultado FDE é que se deve realizar retestes ou reamostragens (como por exemplo, erros na diluição e mau funcionamento de um instrumento).
Esses testes devem fazer parte da investigação do laboratório e devem ser efetuados para confirmar ou descartar uma possível causa raiz, dessa forma devem envolver apenas as soluções originais.
Não é aceitável efetuar novos testes sem uma hipótese documentada com intenção aleatória de achar a causa.
Plano de reanálise, reteste ou reamostragem

Deve haver um plano para as reanálises, retestes ou reamostragens realizadas nas Fases I e II, para os casos em que a causa raiz do resultado FDE não tenha sido identificada.
O plano deve envolver:
- Descrição desses novos testes;
- Aprovação da GUIA Nº 8, VERSÃO 1, de unidade/garantia da qualidade antes do início da investigação;
- Detalhamento da hipótese que originou os novos testes;
- Definição de quais amostras serão retestadas;
- Procedimento dos testes e como os dados serão tratados e avaliados.
Essa parte da investigação (reanálises/retestes/reamostragens) não pode ser usada para substituir resultados analíticos originais, mas somente para confirmar ou descartar uma provável hipótese.
Reanálise ou Reteste

O reteste ou reanálise, pode ser considerado caso não tenham sido identificadas falhas no laboratório ou a causa raiz do resultado FDE durante a investigação nas etapas de produção. Quando disponível, a amostra utilizada para o reteste deve ser a mesma que originou o resultado FDE.
O reteste deve ser realizado quando a investigação indica mau funcionamento em determinado equipamento/instrumento ou para identificar problema no manuseio da amostra, como por exemplo, um erro na diluição.
É importante considerar no plano de reteste a inclusão de um analista diferente daquele que efetuou o teste original e possuir, no mínimo, qualificação e experiência semelhante no teste em questão. Não é aceitável a execução de retestes até um resultado “dentro da especificação” ser obtido, descartando-se desta forma o resultado FDE sem justificativa.
O número máximo de retestes a serem realizados em uma amostra deve estar em procedimento e sempre deve ser baseado em princípios técnicos estatisticamente válidos (alguns guias sugerem 5, 7 ou 9 vezes). O número de retestes não pode ser reajustado (ser diferente do procedimento ou do plano).
No caso de um erro no laboratório tenha sido identificado, os resultados do reteste irão substituir o resultado original, que deve ser rastreável.
Caso não tenha sido identificado erro no laboratório, não há razão para os resultados dos retestes substituírem o resultado original. Todos os resultados devem ser relatados e considerados nas decisões finais quanto à disposição do lote.
Reamostragem

A reamostragem deverá raramente ser efetuada uma vez que novas variáveis serão introduzidas na investigação. Esta deverá ser tecnicamente embasada e formalmente aprovada pela Garantia da Qualidade.
Enquanto o reteste se refere a uma nova análise da amostra original, a reamostragem envolve outras amostras utilizando-se unidades tomadas por meio do plano de amostragem original. Uma nova amostragem no lote pode ser necessária uma vez que pode ter sido identificada falha na amostragem original.
Considerações sobre ensaios microbiológicos

É importante avaliar as condições dos testes cuidadosamente e determinar quais são os limites entre as áreas de amostragem e manufatura. A definição deve ser estabelecida para se determinar se um ou mais lotes foram impactados.
Inicialmente não se deve descartar nenhuma possibilidade de causa raiz. Nessa etapa é recomendado que se utilize ferramentas da qualidade para auxiliar no levantamento das possíveis causas raízes (Brain storm, Ishikawa, etc). Assim deve-se incluir, dentre outros:
- A definição de uma hipótese a ser investigada;
- A identificação do microrganismo e as suas possíveis fontes de contaminação e suas compatibilidades com os tipos de microrganismos encontrados. As identificações devem ser em nível de DNA/RNA;
- A confiabilidade do fornecedor das cepas padrões de microrganismos;
- A qualidade do meio de cultura, seu método de preparação, fornecedor, histórico de esterilização;
- Status de validação, qualificação e limpeza dos equipamentos e utilidades utilizadas;
- Resultados e dados de tendência do monitoramento ambiental das áreas de teste e de suporte;
- Limpeza e manutenção dos ambientes de testes;
- Dados de eficácia dos desinfetantes utilizados;
- Eventos não usuais que podem ter potencial impacto na análise;
- Os registros de desvios e de intervenções de manutenção.
Devido à variabilidade dos resultados microbiológicos, não se deve limitar a investigação a um lote específico. A ampliação da revisão incluindo os dados de lotes anteriores e posteriores deve ser considerada. Especial atenção deve ser tomada sobre os lotes posteriores, que por ventura podem apresentar tendência de resultados FDE e evidenciar desvios de qualidade maiores.
Considerações sobre Estudo de Estabilidade

Os resultados FDE envolvendo estudo de estabilidade deverão prontamente ser investigados. Pode-se utilizar o mesmo fluxo de entendimento desse guia. Casos de resultados fora de tendência não serão cobertos por esse guia.
Fase III
Caso o lote seja rejeitado ainda é necessária investigação para determinar se outros lotes foram afetados e abertura de CAPA. Nessa fase é feito o relatório final de investigação. Deve haver a revisão completa da investigação efetuada na produção e no controle de qualidade.
Para a conclusão da investigação todos os resultados devem ser avaliados, deve haver um relatório contendo um sumário das investigações efetuadas e uma conclusão detalhada. Caso não tenha sido possível a determinação da causa raiz, uma causa raiz mais provável pode ser sugerida.
O impacto do resultado FDE em outros lotes, nos estudos de estabilidade que estão sendo conduzidos, na validação do processo e nos procedimentos de análise deve ser avaliado pelo controle de qualidade e pela Garantia da Qualidade. Ações corretivas e preventivas apropriadas devem ser adotadas e deve ser definida a disposição do lote.
Caso o lote seja rejeitado não há limite para realização de outros testes que objetivem a determinação da causa raiz, de forma que ações corretivas possam ser implementadas. Todavia a decisão de rejeição de um lote não pode ser revertida com a justificativa de resultados de novos testes.
Interpretação dos Resultados

A Garantia da Qualidade é responsável pela interpretação dos resultados da investigação, deve utilizar uma abordagem estatística para se determinar o resultado da investigação. A abordagem depende do tipo de teste, da amostra e do objetivo da amostragem.
É necessário ter cautela com o uso de valores médios, pois além de mascararem a variação dos resultados individuais, o valor médio poderá atender as especificações e conduzir a uma interpretação errônea do resultado final. Desta forma, no contexto da fase de investigação do resultado FDE não se deve utilizar a média dos resultados originais, dos retestes ou mesmo dos resultados dos testes obtidos na nova amostragem.
Não é usual que, numa série de análises com métodos validados, os resultados sejam muito diferentes entre si. Caso isso aconteça, estes valores são considerados discrepantes, podendo ser derivados de desvios dos métodos descritos ou resultantes da variabilidade da amostra. Um resultado inicial FDE não necessariamente significa que o lote falhou e deve ser rejeitado. Deve haver uma investigação es seus resultados, que podem incluir resultados de retestes e reamostragens, devem ser interpretados sob a ótica da análise de risco para avaliação sobre a disposição do lote.
Quando uma causa raiz é encontrada e o resultado FDE é invalidado, o resultado não deve ser usado para avaliar a qualidade do lote. A invalidação de um resultado somente pode ser efetuada com todas as observações documentadas que provam que o resultado não era válido.
Nos casos que o resultado FDE é confirmado, deve haver avaliação da qualidade do lote do produto para a sua rejeição. Dessa forma a investigação de resultado FDE deve ser ampliada para a investigação de falha do lote, devendo ser estendida a outros lotes e outros produtos que possam estar associados a esta falha.
Nos casos de investigação inconclusiva, ou seja, sem determinação da causa raiz na Fase I e Fase II, não há base científica para invalidar o resultado FDE e afirmar que o teste apresenta resultados satisfatórios. Igualmente, um resultado FDE encontrado na primeira análise não necessariamente significa que o lote foi reprovado e deve ser rejeitado.
[quote_center]Todo resultado FDE deve ser investigado e registrado, incluindo neste âmbito os dados da investigação da Fase I e II para uma decisão sobre liberação do lote com uma análise robusta do cenário.[/quote_center]
Esta análise deve considerar:
- Se os resultados foram atípicos e os retestes robustos;
- Se as investigações no laboratório e na produção não indicaram eventos e nenhuma variação que pudessem indicar falhas;
- Se o histórico do processo e do produto não demonstrou tendências;
- Se todos os resultados estatisticamente válidos dos retestes estavam dentro da variabilidade do método utilizado (e dentro da especificação).
No mesmo cenário, quando a investigação é inconclusiva, ou seja, a causa raiz não foi encontrada e as hipóteses levantadas não confirmaram o resultado FDE, deve ser determinada uma causa mais provável pela Garantia de Qualidade.
Qualquer decisão que opte pela liberação do lote para a comercialização, deve ocorrer somente após uma investigação robusta e que demonstre que o resultado FDE não afeta a qualidade do produto. Os resultados de monitoramento de controles em processo, dentre outros, podem levar à conclusão de que o resultado FDE não reflete a real qualidade do lote. No caso de dúvidas (conclusões e resultados não robustos) deve-se sempre considerar o princípio da precaução e o lote ser rejeitado.
Conclusão e download dos Guias

Bom o texto é longo, mas necessário, visto a complexidade das informações e detalhes da execução dos estudos investigacionais.
E para ajudar, e até para que tenha mais detalhes sobre como proceder, trouxemos os dois guias aqui para download:
FDA: OOS Investigations Guideline 2006
ANVISA: Guia de Investigação de Resultados Fora de Especificação
E lembre-se, se precisar de suporte técnico, ou mesmo de mão-de-obra para elaborar os relatórios da sua empresa conte com a ajuda da equipe da Consultoria Farmacêuticas!
Consultoria Farmacêuticas

Fernanda de Oliveira Bidóia
Diretora Técnica
Telefones:
Comercial + 55 11 3392 2424
Celular: + 55 11 992961326
End: Av. Marquês de São Vicente, 446 cj 1102
São Paulo – SP
http://consultoriafarmaceuticas.com.br/
Abraços a até o próximo artigo!
Anna Majewski e Fernanda de Oliveira Bidoia
Fonte:
Guia nº 8/2017 – Investigação de Resultados Fora da Especificação (FDE).
Investigating Out-of-Specification (OOS) Test Results for Pharmaceutical Production.





Anna e Fernanda, boa tarde!
Como estão?
Tenho uma dúvida… Quais guias existem que possam ajudar na determinação do número de retestes após obtenção de um resultado fora da especificação? O número de retestes precisa do embasamento estatístico ou apenas realizando a troca do analista e/ ou a comparação da triplicata já seria o suficiente para justificar os meus retestes com base em um racional?
Obrigado,
Lucas Prado
Oi, Lucas!
Segundo o guia da Anvisa de resultados Fora da Especificação – FDE, “O reteste, ou reanálise, pode ser considerado caso não tenham sido identificadas falhas no laboratório ou a causa raiz do resultado FDE durante a investigação nas etapas de produção. Quando disponível, a amostra utilizada para o reteste deve ser a mesma que originou o resultado FDE. Para compostos líquidos, a amostra para reteste pode ser tomada da amostra original do produto e para compostos sólidos, pode ser uma quantidade adicional tomada da amostra preparada para o teste originalmente”.
O guia ainda fala que: “Não é aceitável a execução de retestes até um resultado “dentro da especificação” ser obtido, descartando-se desta forma o resultado FDE sem justificativa. O número máximo de retestes a serem realizados em uma amostra deve estar em procedimento e sempre deve ser baseado em princípios técnicos estatisticamente válidos (alguns guias sugerem 5, 7 ou 9 vezes). O número de retestes não pode ser reajustado (ser diferente do procedimento ou do plano).”
No entando, ao meu ver, mais que dois retestes deve ser questionável.
A referência em questão está no próprio guia: INVESTIGAÇÃO DE RESULTADOS FORA DE ESPECIFICAÇÃO – FDE – ANVISA
Espero ter respondido à sua dúvida.
Abs