
Questionamentos da Validação de Limpeza:
Quando um equipamento limpo está suficientemente limpo?
De que maneira científica pode ser comprovada uma limpeza?
Qual o limite de aceitação de resíduos?
Como este limite é determinado?
Estas são as principais questões a serem definidas e comprovadas em uma validação de limpeza.
Neste post, os assuntos relacionados à documentação não serão o foco, pois é fundamental que se tenha os 6 principais documentos:
- Análise de risco ou Grid System do ativo de escolha (mais crítico).
- Validação dos métodos analíticos do ativo, detergente, produtos de degradação (quando descrito em farmacopeia) e microbiológico.
- Procedimento de limpeza do equipamento aprovado e implementado.
- Protocolo de validação de limpeza aprovado do equipamento no qual será submetido a validação limpeza.
- Cálculos para determinação dos limites físico-químicos dos ativos, detergentes e limites microbiológicos.
- Relatório.
Em outro post serão discutidos assuntos relacionados aos itens necessários na documentação de validação de limpeza.
Então, vamos ao passo-a-passo da execução da validação de limpeza:
-
Escolha do ativo mais crítico na validação de limpeza
Faça a análise de risco ou um Grid System e escolha o ativo/produto mais crítico de acordo com o seguintes critérios:
- Classe terapêutica
- Toxicidade
- Atividade farmacológica do produto anterior,
- Produtos secundários ou produtos de degradação;
- Solubilidade em água
- Dificuldade de limpeza
- Produto Controlado (Portaria nº 344) – apenas para indústria farmacêutica
- Dose terapêutica máxima diária do próximo produto – apenas para indústria farmacêutica
- Possibilidade de crescimento microbiológico;
Faça uma análise com todos os dados de ativos que passam pelo equipamento e anexe à documentação de validação.
[quote_center]Para cosméticos e saneantes, outros critérios devem ser avaliados, tais como: classificação do produto (cosmético em grau 1 e 2; saneantes segundo classificação da ANVISA), presença de pigmentos de difícil remoção, alergenicidade, e veículo utilizado, por exemplo.[/quote_center]
-
Validação da metodologia analítica
Obrigatoriamente os métodos analíticos devem estar validados antes da validação de limpeza do equipamento ser iniciada.
A numeração/códigos dos relatórios de validação analítica devem ser mencionados no protocolo de validação de limpeza, incluindo a versão e data de aprovação.
[quote_center]A validação do método analítico deve ser realizada juntamente com os testes de recuperação.[/quote_center]
O método analítico deve ser desafiado em combinação com o método de amostragem selecionado, comprovando que o contaminante pode ser recuperado da superfície do equipamento e qual o seu nível de recuperação.
Deve-se determinar a porcentagem de recuperação, bem como os parâmetros de metodologia analítica (especificidade, linearidade, intervalo, precisão, limite de detecção, limite de quantificação e exatidão do método).
Métodos que devem estar obrigatoriamente validados:
- Ativo/produto
- Produto de degradação (RDC 58/2013)
- Microbiológico
- Análise da água potável
- Análise da água PW
- Detergentes
- Agentes saneantes
-
Limpeza visual

Durante a execução de uma validação de limpeza este é o primeiro item a ser verificado. Caso seja detectado que o equipamento está sujo este é imediatamente reprovado, sem discussão.
Mas se este é considerado limpo, qual o limite de resíduo não detectado visualmente?
Para mais informações sobre este item, leia o artigo “Como determinar o visualmente limpo na validação de limpeza?”
-
Determinando o limite no produto subsequente (LSP – Limit in Subsequent Product)
O ponto inicial para determinação do limite de aceitação é o quanto de resíduo de um produto poderá passar para o lote subsequente, após a limpeza de um determinado equipamento, sem que haja risco de contaminação a nível prejudicial para a saúde de um paciente.
Para tanto, o FDA e a Anvisa determinam:
- O equipamento deve estar visualmente limpo
- Qualquer princípio ativo pode estar presente no produto subsequente num nível máximo de 10 ppm
- Qualquer princípio ativo pode ter no máximo de 1/1000 ppm da dose mínima diária (resíduo) dentro da dose maxima diária do produto subquente.
Complicado? Vamos ao desenho:
Produto A (mais crítico) – ativo de escolha da validação
Produto B – produto subsequente
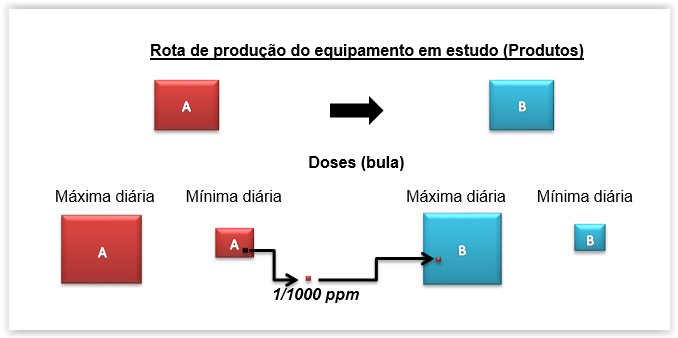
Agora ficou mais fácil compreender. Certo?
Então vamos ao próximo item a ser avaliado:
-
Limite por área de superfície
Isso realmente pode gerar dúvidas…
Mas aqui estão dois itens de extrema importância:
1º – Qual limite deve ser usado?
O limite depende do LSP (Limit in subsequent product), ou seja, deve ser levado em consideração o menor valor: 10 ppm ou o valor encontrado atraves do cálculo de LSP.
2º – Qual a área a ser considerada no estudo?
Para este estudo, deve ser levado em consideração a área de superfície do equipamento em cm2 que entra em contato diretamente com o produto.
Para o cálculo detalhado veja o post: Desvendando o cálculo de LeBlanc na validação de limpeza
Mas há um detalhe muito importante: para o cálculo deve ser utilizado não somente a área do equipamento em cm2, e sim toda a área da rota de produção.
Resumindo: use o valor obtido (cm2) na soma de todas as áreas de todos os equipamentos que entram em contato com o ativo de maior criticidade durante a etapa do processo de fabricação.
O estudo, levando em consideração a área total de superfície (rota), é importante para determinação de um limite residual baixo. Desta forma, o critério de aceitação do resíduo torna-se muito mais rígido.
[quote_center]Nota: Esse item é amplamente avaliado pela ANVISA.[/quote_center]
Fica a dica.
-
Limite na análise da amostra
Agora que ficou claro a questão da rota de produção, de que maneira será determinado qual é o produto subsequente mais crítico para ser usado no estudo?
Para tanto deve ser feita uma análise combinatória dos critérios descritos abaixo para avaliar qual o menor resultado. O menor resultado, ou mais crítico, é o produto subsequente de escolha a ser considerado no cálculo:
- Relacione todos os produtos que passam no equipamento a ser validado
- Descreva a dose máxima e mínima (em mg) de cada substância ativa
- Coloque o tamanho do lote de cada produto (em Kg)
- Coloque a rota de produção de cada produto em cm2
- Faça o cálculo do limite para cada um dos produtos subsequentes
- Selecione o mais crítico, ou seja, o de menor valor.
-
Área de amostragem
Para realizar o cálculo do limite, e também para a amostragem durante a execução da validação de limpeza, é necessária a determinação da área de amostragem.
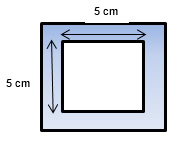
LeBlanc utiliza em seu cálculo uma área de 25 cm2.
Para tanto, é necessário usar um gabarito de plástico descartável com 5 cm de cada lado interno (5 x 5 cm) para totalizar a área de 25 cm2. Éssa medida é perfeita para amostrar nos pontos considerados mais críticos, em termos de acúmulo de resíduos, no equipamento (definidos em protocolo).
Há laboratórios que utilizam uma área maior, de 100 cm2 (10 x10 cm de gabarito). Sinceramente, acho uma área muito grande.
Levando em consideração que todo estudo é baseado em seguir o menor limite, porque ampliar toda área? Acho que tal aumento deve ser muito bem justificado, principalmente diante de uma inspeção da ANVISA.
É logico que existem exceções, como é o caso de produtos hormonais que as dosagens são muito baixas e, consequentemente, os limites encontrados dificilmente serão detectados através dos equipamentos de análise disponíveis no laboratório de Controle de Qualidade. Mesmo neste caso, o aumento da área deve ser muito bem justificado em protocolo e relatório de validação.
-
Amostragem
Escolha os pontos mais críticos do equipamentos, ou seja, de maior dificuldade de limpeza.
Defina com critério qual o melhor método de amostragem a ser usado para cada tipo de limpeza:
- Amostragem direta
- Amostragem indireta
A combinação dos dois métodos, em muitos casos, é recomendado.
Método direto (Swab ou esfregaço)
Deve ser feita com gabarito de 5 x 5 cm nos pontos de maior dificuldade de limpeza do equipamento. Todos os pontos definidos em protocolo.
Devem ser amostrados e analisados:
- Resíduos de ativo
- Resíduos dos agentes de limpeza (detergente)
- Possível contaminação microbiana (contagem total de bactérias e pesquisa de agentes patógenos)
O plano de amostragem deve ser elaborado em conjunto com o Controle de Qualidade e deve constar no protocolo de validação.
Para a amostragem:
- Utilize swab
- Solventes utilizados na amostragem devem ser definidos pelo Controle de Qualidade, de acordo com o ativo e os detergentes em estudo.
- Gabarito
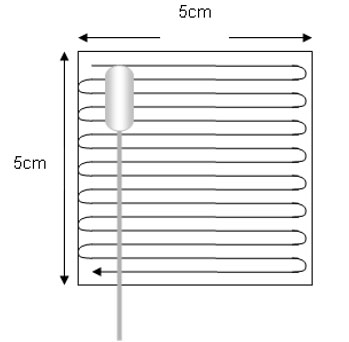
- Sentido da amostragem micro

- Sentido da amostragem do ativo e detergente
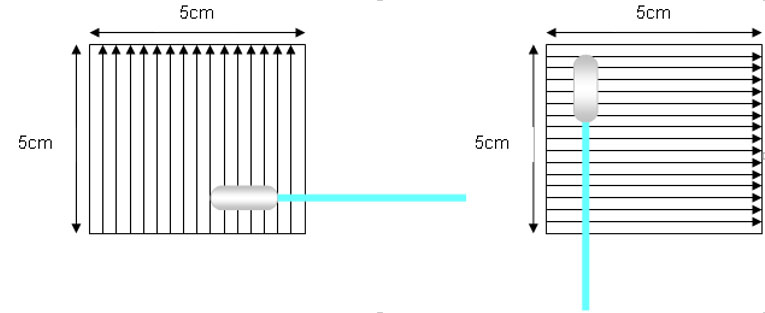
- Quantidade de amostras definida pelo CQ
- Número de limpezas: no mínimo 3 limpezas completas de 3 lotes consecutivos
Método indireto (rinsagem)
Há outras técnicas de amostragem, como a rinsagem, por exemplo. Neste caso, determine o volume e tipo de solvente a ser empregado (rinsagem).
Para saber mais sobre o método leia o post Uso de amostragem por rinsagem na validação de limpeza
-
Fator de correção
Insira um fator de correção no cálculo de acordo com o produto em questão e a também de acordo com a fator de recuperação do swab.
O fator de recuperação deve ser determinado pela equipe de validação analítica.
10. Análise de resíduo de detergente
O limite para o cálculo de resíduo de detergente pode ser feito através do cálculo de NOEL.
Cálculo do NOEL (Nível de Efeito Não Observado): determina a concentração segura do total de resíduos, no caso, do agente de limpeza.
Os resultados obtidos das amostras analisadas devem ser divididos pelo fator de recuperação do swab, e devem ter valores menores que o resultado teórico para passarem no critério de aceitação.
11. Contaminação microbiológica
Determine um critério de aceitação para o limite microbiano.
Faça testes para contaminação microbiana (contagem total de bactérias) e pesquisa de agentes patógenos.
O plano de amostragem deve ser elaborado pelo laboratório de Microbiologia. Este, assim como o solvente (soro fisiologico), e o critério de aceitação, devem estar relacionados em protocolo.

Dica: [quote_center]Testes indiretos, tais como condutividade e TOC, são importantes para o monitoramento da rotina de limpeza dos equipamentos , mesmo que este tenho sido validado. [/quote_center]
Esses são os principais passos para executar uma validação de limpeza perfeita e ainda arrasar na inspeção da ANVISA.
Mas se ainda tiver dúvida mande um e-mail para contato@farmaceuticas.com.br ou adquira o curso completo de validação de limpeza:
Curso de Validação de Limpeza
Caso necessite de aprimoramento ou queira se aprofundar nas técnicas e tipos diferentes do estudo, faça o Curso online de Validação de Limpeza.
Elaborado por profissional com mais de 14 anos de experiência na atividade, o curso traz técnicas, critérios, cálculos e sistemáticas atuais utilizadas internacionalmente, e que também atendem às Normas da ANVISA.
Confira!!!

https://cursosfarmaceuticas.com.br/produto/curso-de-validacao-de-limpeza/
Consultoria especializada em Validação de Limpeza
Fernanda de Oliveira Bidóia
Consultora de SGQ, Validação e Qualificação
Consultoria Farmacêuticas
Av. Marquês de São Vicente, 446 cj 1102
São Paulo – São Paulo
Tel: 55 11 3392 2424
cel: 55 11 99296 1326
fernanda@farmaceuticas.com.br
contato@farmaceuticas.com.br
www.farmaceuticas.com.br
Referência
FDA, “Guide to Inspections of Validation of Cleaning Processes”
PDA, Cleaning Validation
D.A. LeBlanc, “Rinse Sampling for Cleaning Validation Studies”
PIC: Principles of Qualification and Validation in Pharmaceutical Manufacture – Recommendations on Cleaning
Validation. (ref. Document PR 1/ 99 March 1999)
ANVISA: Validação de Limpeza para Farmoquímicos





[…] 11 Passos para a execução da validação de limpeza […]
[…] Para mais informações sobre o assunto leia o post 11 Passos para a execução da validação de limpeza. […]
[…] Validação de limpeza (total de equipamentos, em campanha, utensílios, salas produtivas e vestimentas, validação do tempo de limpo e validação do tempo de sujo do equipamento); […]